在外部环境改变时,蛋白质的空间构型会发生变化,由可溶的天然态转变为不溶的纤维态,该过程被称为蛋白质的淀粉样纤维化( Amyloidosis ) 。 [ 1-3 ] 蛋白质的淀粉样纤维化过程起始于蛋白分子的错误折叠,这些错误折叠首先生成寡聚体( oligomers ),继而叠加形成原纤维( protofibrils )并最终自组装形成纤维或纤维聚集体( fibrils )。 [ 4 ] 蛋白质的纤维化过程最早发现于某些疾病的病变组织中,是帕金森症、阿尔兹海默症、 Ⅱ- 型糖尿病等多种淀粉样变性疾病的重要致病原因。根据 2014 年的统计数据,淀粉样变性疾病已成为美国老年人中的第三大致死疾病,对人类健康和社会经济带来了巨大的威胁。在蛋白质的生产和储运过程中,尤其是蛋白质的药用过程中,淀粉样纤维化过程会导致免疫原性,使病人产生不同程度的不适反应。因此,对蛋白质淀粉样纤维化过程的研究,例如其纤维化机理及抑制方法等,对于淀粉样变性疾病的预防、治疗、生物技术的发展以及药物开发等领域都有重要意义。
近年来,“淀粉样纤维化是蛋白质的普遍性质”逐渐受到人们的认可。除膜蛋白和纤维蛋白外,几乎所有蛋白质都能在一定条件下自组装形成纳米纤维结构。在自然界中,蛋白质纳米纤维作为一维纳米材料,借助于优良的力学性能和化学活性成为诸如骨骼、皮肤、血管等多种生物组织中不可或缺的构筑单元。受此启发,越来越多的研究着眼于无生物毒性蛋白质纳米纤维的实验室制备,即通过改变实验条件( pH 、温度、离子强度等),诱导或促进蛋白质自组装形成纳米纤维。这些实验室制备的一维蛋白质纳米纤维没有生物毒性、长径比高、力学性能优异且表面基团丰富。以蛋白质纳米纤维作为结构基元,利用其表面荷电性、亲疏水性等特点,通过静电吸附、疏水力、氢键及物理(化学)交联等分子间作用力可构筑一维(纤维)、 [ 5 ] 二维(片、膜) [ 6 ] 及三维(凝胶) [ 7-9 ] 功能材料,在诸如组织支架、生物传感器、催化、光电材料、仿生材料以及许多新兴材料等领域都有很高的应用潜力。因此,蛋白质纳米纤维的制备与应用对于生物材料、纳米材料等领域同样具有积极的推动作用。
1. 1 蛋白质 纳米纤维的研究意义
1. 1 .1 病理学研究
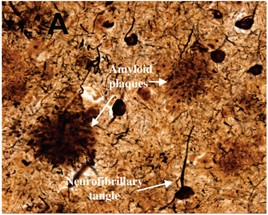
蛋白质淀粉样纤维化是很多疾病的诱导因素,例如帕金森症、阿尔兹海默症、 Ⅱ- 型糖尿病等。 [ 4 ] , [ 10 , 11 ] 蛋白质的淀粉样变性通常形成稳定、高长径比的一维纳米纤维结构。这些纤维可与细胞膜相互作用导致细胞损害或者改变细胞的生理生化途径进而诱导细胞凋亡。以阿尔兹海默症为例,如图 1.1 所示,蛋白质淀粉样纳米纤维在神经组织中堆积形成斑块和缠结,破坏组织功能,继而引发疾病。 [ 12 ]
图 1 .1 阿尔兹海默症组织中的缠结和斑块 [ 12 ]
蛋白质淀粉样纤维化的致病机理目前尚不明确,这源于其过程的复杂多样性。例如,不同疾病中蛋白质淀粉样纤维化的形式不同,其发生具有不确定性,有些甚至具有可遗传性;蛋白质淀粉样纤维在组织中的堆积可发生在细胞内亦可发生在细胞外。蛋白质淀粉样变性过程中通常包含四种典型结构,即单体( monomer )、寡聚体( oligomer )、原纤维( protofibril )和纤维( fibril )。成熟的蛋白质淀粉样纤维通常具有生物毒性,早期的研究认为组织中巨大的纤维沉积损害细胞活性并诱发相关的疾病。近期的一些研究则认为作为中间产物的寡聚体拥有比成熟纤维更高的生物毒性。 Meenakshi Verma 等人基于帕金森症和阿尔兹海默症系统描述了蛋白质淀粉样纤维化过程并讨论了不同产物的生物毒性 差异(图 1.2 )。 [ 13 ] 他们认为寡聚体的生物毒性高于成熟纤维,并从二者的结构差异角度做出解释。主要表现为以下四点:
1 :寡聚体 β - 折叠结构中的疏水面暴露在外而成熟纤维结构中的疏水面则隐 藏在内部堆叠结构中。 [ 14 ]
2 :寡聚体相较于成熟纤维尺寸更小,更容易进入组织中。 [ 15 ]
3 :寡聚体具有更多的活性位点,更容易与细胞反应。
4:成熟纤维相较于寡聚体更加稳定。
图 1. 2 蛋白质淀粉样纤维化聚集示意图。四种不同的聚集方式分别为:( A )蛋白质错误折叠使空间构型改变形成原纤维继而形成纤维。( B )错误折叠的蛋白质单体形成寡聚体但不形成纤维。( C )成熟纤维剪切或分裂形成纤维状寡聚体后重新形成纤维。( D )成熟纤维作为模板使单体形成可分散的寡聚体并催化二次成核反应 [ 13 ]
为了预防和治疗蛋白质淀粉样纤维化疾病,越来越多的研究着眼于 寻找可抑制蛋白质淀粉样纤维化过程的物质,预防疾病的发生。例如,一些小分子可通过调控蛋白质淀粉样纤维化过程能垒,使蛋白质维持在原始状态,进而预防淀粉样纤维化疾病的发生。 [ 16 ] 除此之外,一些蛋白质(如 α B- 晶状体蛋白)、 [ 17 ] 多酚类、 [ 18 ] 有机染料 [ 19 ] 等均可不同程度地抑制蛋白质纤维化过程。近年来,由于特效药物的缺乏,越来越多的研究将目光转向人类的生活及饮食习惯对蛋白质淀粉样纤维化过程的影响。例如,根据 2006 年的一篇报道,地中海饮食结构(橄榄油比例高)可抑制阿兹海默症的发生。 [ 20 ] 深入了解不同因素,尤其是和人体相关的环境因素对蛋白质纤维化过程的影响机制,有效抑制蛋白质淀粉样纤维化过程,是一种合理易行、创伤小的疾病防治手段。
1. 1 .2 材料学研究
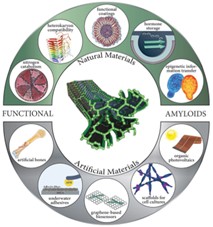
除上述病理性蛋白质外,非致病 蛋白质自组装纳米纤维具有良好的生物相容性,而且密度低、透明性高、力学性能优良,在薄膜、多孔支架、凝胶、微米级纤维等多种功能材料的制备中都发挥重要作用。[ 21-23 ] 除此之外,蛋白质自组装纳米纤维含有多种氨基酸残基,这些氨基酸残基具有不同的结构、极性以及荷电性,使得蛋白质纳米纤维具有双电层结构并同时具备亲水区域和疏水区域。加之蛋白质纳米纤维长径比高的结构特性,使其作为结构基元可通过静电力、疏水力、氢键、 π-π 相互作用等分子间作用力自身或复合其它纳米材料构筑一维、二维及三维功能纳米材料。这些材料可用于诸多领域,例如药物释放、光伏电池、生物传感器、发光二极管、电化学、生物探针、细胞培养等(图 1.3 )。 [ 24 , 25 ] 例如,源于小鼠层粘连蛋白的蛋白质纳米纤维作为细胞支架通过结合活性生物分子可以控制细胞生长; [ 26 ] 葡萄糖氧化酶表面修饰的乳清蛋白自组装纳米纤维可通过分子中的巯基吸附在金基底上,并作为生物传感器检测葡萄糖。 [ 27 ]
图 1. 3 蛋白质淀粉样纳米纤维应用多样性示意图 [ 25 ]
1. 2 蛋白质纤维化过程及结构表征
1. 2 .1 生物体中的蛋白质纤维化
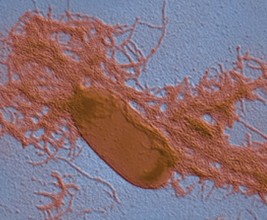
在自然界中,蛋白质分子的纤维化自组装除与多种疾病相关,还与许多特定生物功能的实现相关,例如催化、信息存储以及组织保护等。 [ 28-30 ] 蛋白质自组装纳米纤维具有出色的稳定性,对许多化学或生理条件都有很高的耐受性,使其在生物体中多作为结构材料以达到保护生物体的作用。例如,许多细菌的生物膜中都含有蛋白质纳米纤维成分,如大肠杆菌、沙门氏菌、枯草杆菌、假单胞菌、葡萄球菌等。 [ 31-35 ] 图 1.4 所示为大肠杆菌生物膜中的蛋白质纳米纤维电子显微镜照片。 [ 33 ] 值得注意的是,不同于许多病理性蛋白质的纤维化过程,细菌生物膜中的纤维化过程经过精确的控制,既保证蛋白质纤维化过程的正常进行,同时也确保细胞功能不受影响。除单细胞生物外,蛋白质纳米纤维作为结构材料同样可见于许多昆虫的蛋壳中。例如,蚕茧中含有大量蛋壳绒毛蛋白,其中大部分以蛋白质纳米纤维形式存在,可以保护壳内组织免受外部环境的侵害。 [ 36 ]
图 1. 4 大肠杆菌细胞的电子显微镜照片。大肠杆菌细胞产生蛋白质淀粉样纳米纤维并协助生物膜的形成 [ 33 ]
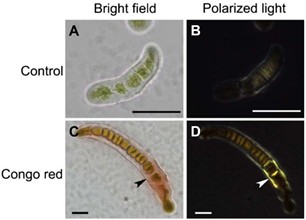
蛋白质自组装纳米纤维长径比高,同时具备亲水性和疏水性,使蛋白质纳米纤维对多种表面都展现出良好的黏附性 , 包括气 - 液界面、油 - 水界面、脂质膜 - 水界面、以及固 - 液界面等。 [ 37-39 ] 例如,生物体中,一些蛋白质自组装纳米纤维在液体界面的缔合能可达约 6000 KB T , [ 37 ] 使得纤维在界面的吸附过程不可逆,类似于纳米颗粒对皮克林乳液的稳定过程。一些陆生藻类中提取的黏附物质中,蛋 白质纳米纤维亦为主要成分(图 1.5 )。 [ 40 ]
图 1. 5 溪藻属铁芒萁黏附剂中蛋白质淀粉样纳米纤维刚果红双折射表征。未染色的藻类作为对照在( A )明场及( B )正交偏光下照片,展现出微弱的自发荧光;刚果红染色处理的藻类在( C )明场中的照片展现出藻类复叶处黏附区域与刚果红染液的结合,( D )相应区域在正交偏光下展现金绿色双折射光,说明淀粉样纳米纤维的存在。标尺: 25 微米 [ 40 ]
蛋白质纳米纤维除了在单细胞生物(细菌、真菌)中具有生理作用,在人体中同样发挥重要作用。例如,人体中的蛋白质 pmel17 可以自组装形成纳米纤维催化支架,并有效催化醌前驱体形成黑色素。 [ 41 ] 该过程中蛋白质纳米纤维催化支架包裹有毒的黑色素前驱体并防止其扩散和损害组织细胞。蛋白质 pmel17 纳米纤维催化支架的生物惰性、高比表面积及催化活性是其完成该生理过程的基础。近期的研究还发现蛋白质纳米纤维出色的稳定性使其可作为人体中激素的存储仓库,并可调控激素的释放。 [ 42 ]
1. 2 .2 实验室中的蛋白质纤维化
受 生物体中蛋白质纤维化过程的启发,许多非致病蛋白质( 如 玉米醇溶蛋白、 [ 43 ] 小麦谷蛋白、 [ 44 ] 溶菌酶、 [ 45 ] β - 乳球蛋白、 [ 46 ] 蚕丝蛋白 [ 47 ] 和多肽( Aβ (1-42) ) [ 48 ] 等 ) 在不同的实验条件可自组装形成纳米纤维。蛋白质分子中的氨基酸残基通过分子间氢键、疏水力等作用使蛋白质分子维持精密的空间构型。外部环境的改变可以影响蛋白质分子的荷电性和亲疏水性,例如 pH 接近等电点时,蛋白质分子不带电;溶液离子强度的改变可以屏蔽蛋白质分子中的静电排斥作用,不良溶剂的加入可以促使蛋白质分子中疏水区域外露。这些改变有利于蛋白质分子内的疏水力相互作用,并诱导蛋白质分子自组装形成高长径比的纳米纤维结构。基于不同的驱动力,多种蛋白质自组装纳米纤维成功实现实验室制备。例如, β - 乳球蛋白在 pH=2 、 80 °C ,低离子强度 ( I ≤ 20 mM ) 条件下可形成高长径比的纳米纤维(图 1.6 A ); [ 49 ] 牛血清白蛋白( BSA )在磷酸盐缓冲液( 50 mM 、 pH=7.4 )、 60 °C 及乙醇( 60 vol.% )存在条件下可形成蠕虫状纳米纤维( 图 1.6 B ); [ 50 ] 溶菌酶在 pH=2 、 60 °C 、缓慢搅拌条件下可形成无分支、刚性纳米纤 维(图 1.6 C )。 [ 51 ] 蛋白质自组装纳米纤维形成过程为动力学控制过程,通常经过诱导成核期后纤维迅速生长并达到稳定。不同蛋白质其诱导期不同,如图 1.6 D 所示,预先加入成核位点作为“种子”可有效缩短诱导期。 [ 52 , 53 ]
图 1. 6 ( A ) β - 乳球蛋白、( B )牛血清白蛋白、( C )溶菌酶纳米纤维透射电镜图片。( D )蛋白质淀粉样纤维化过程动力学模型 [ 52 ]
1. 2 . 3 蛋白质纤维化 自组装影响因素
蛋白质纤维化自组装过程受诸多因素影响,如体系温度、金属离子、不良溶剂、界面等。 一些外部固体界面的引入可以调控蛋白质淀粉样纤维化过程的成核,进而影响淀粉样纤维化过程。例如, CdTe 纳米颗粒、 [ 54 ] 氟化纳米粒子以及阳离子聚苯乙烯小球 [ 55 ] 与蛋白质单体或寡聚体有很强的结合力,减少了游离单体和寡聚体对蛋白质纤维化自组装成核过程的参与,进而抑制蛋白质淀粉样纤维化过程。 [ 56 , 57 ] 而一些高分子共聚物纳米颗粒、二氧化铈、碳纳米管及二氧化钛纳米颗粒等则可以在其表面富集蛋白质单体及寡聚体,从而加速蛋白质纤维化自组装成核过程,促进蛋白质的淀粉样纤维化。 [ 58-60 ] 金属离子可与蛋白质分子络合进而影响蛋白质的纤维化自组装过程。 [ 61 ] 例如,铜离子可与 β 2 - 微球蛋白结合,引起脯氨酸残基的顺反异构反应,进而促进蛋白质的错误折叠和自组装过程; [ 62 ] 锌离子可与多个 β - 淀粉样蛋白( Aβ )的组氨酸残基络合形成多聚体,这种蛋白质的聚集有利于蛋白质分子间的相互作用,诱发蛋白质纤维化自组装; [ 63 ] 铜离子与 β - 淀粉样蛋白( Aβ )的络合作用则通过改变蛋白质表面的荷电性,使其趋于电中性,分子间作用力增强,从而加速蛋白质纤维化自组装过程。 [ 64 ]
动态界面和金属离子广泛存在于生态系统中,并参与许多生理生化过程。目前,更多的研究集中在固态界面对蛋白质纤维化自组装过程的影响机理,对于更为复杂的动态界面在蛋白质纤维化过程中的作用,研究相对匮乏。近年来,随着蛋白质结构分析技术的发展及其纤维化过程研究的丰富和深入,越来越多的研究期望解释与人体环境息息相关的因素(如动态界面、金属离子等)对蛋白质纤维化自组装过程的影响机理,并以此寻找新的疾病防治手段和纳米纤维制备方法。
1. 2 . 4 蛋白质纳米纤维的结构表征
蛋白质分子由特定的氨基酸序列构成。氨基酸分子通过分子间酰胺键形成多肽链,多肽链因氨基酸残基结构的不同,借助氢键、疏水力及 π-π 相互作用等分子间作用力进一步形成 α - 螺旋、 β - 折叠、无规卷曲、 β - 转角等二级结构。二级结构进一步通过分子间作用力形成稳定的三级结构和四级结构,从而实现蛋白质分子的构象稳定。蛋白质自组装纤维化过程往往伴随着二级结构的变化,其典型特征是二级结构中 β - 折叠结构 的增多。在蛋白质自组装纳米纤维中,大量的 β - 折叠链通过分子间氢键形成 β - 折叠片结构并沿轴向层层堆叠形成自组装纳米纤维结构(图 1.7 )。 [ 65 , 66 ] β - 折叠的片层结构中氢键密集,决定了蛋白质自组装纳米纤 维的精密结构,同时也为蛋白质自组装纳米纤维的表征提供了理论基础。
图 1. 7 蛋白质淀粉样纳米纤维在不同尺度下的结构特征 [ 27 ]
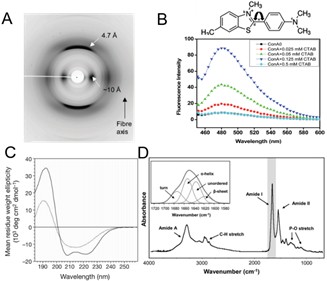
X- 射线散射结果表明,蛋白质自组装纳米纤维包含 4.7 Å 和 10 Å 两个主要的衍射环,分别对应纤维结构中 β ‑ 折叠链间的堆叠距离及 β - 折叠片层间的堆叠距离。(图 1.8 A )。 [ 67 ] 硫磺素 T 染色法是常用的检测蛋白质自组装纳米纤维的手段。硫磺素 T 分子通过与蛋白质 β - 折叠结构结合,使其分子旋转受阻,表现出明显的荧光强度增强( 440 nm 激发, 482 nm 发射)(图 1.8 B )。 [ 68 , 69 ] 圆二色谱( CD )常用于定性及定量表征蛋白质材料的二级结构,如图 1.8 C 所示, α - 螺旋结构在 208 nm 及 222 nm 附近有特征负峰,蛋白质纤维化后于 219 nm 出现单独特征负峰,说明 β - 折叠结构的形成或增多。 [ 70 ] 傅立叶转换红外光谱( FTIR )对于蛋白质二级结构的表征具有操作简便、样品制备简单等优点。通过对蛋白质酰胺吸收带进行去卷积、分峰、拟合等操作可定性、定量分析蛋白质纤维化前后二级结构组成的变化(图 1.8 D )。 [ 71 ]
基于蛋白质自组装纳米纤维结构特征,对于蛋白质自组装纳米纤维的表征通常遵循以下步骤:
① 蛋白质纤维化自组装过程通常伴随体系粘度上升、凝胶化及沉淀等肉眼可见的现象。
② 如出现上述现象,样品可通过硫磺素 T 染色法和刚果红染色法进行测试。
③ 光谱结果初步确认后,可通过透射电镜、扫描电镜或原子力显微镜表征聚集体形貌。
④ 最后通过傅里叶转换红外光谱、圆二色谱、 X- 射线散射等表征其二级结构特征。
图 1. 8 ( A )蛋白质淀粉样纳米纤维的 X- 射线衍射图样,展现出典型的“ cross‑β ”衍射图样。( B )硫磺素 T 结构式(上)、 CTAB 诱导伴刀豆球蛋白 A 所成纳米纤维的硫磺素 T 荧光增强(下)。( C )肌红蛋白及肌红蛋白纳米纤维的圆二色谱。( D )傅里叶转换红外光谱对蛋白质纤维化前后二级结构的表征,包括不同酰胺吸收带以及酰胺 Ⅰ 带不同吸收峰对应的二级结构 [ 71 ]
蛋白质自组装纳米纤维独特的分子结构赋予其出色的材料性能。例如,与其它一维生物高分子材料不同,蛋白质纳米纤维在结构上与传统的带电聚合物类似,其在不同溶剂中的长度恒定。 [ 72 , 73 ] 这得益于蛋白质纳米纤维内密集的氢键作用,使其可有效抗拒溶剂影响及静电排斥作用。 [ 74 ] 另外,蛋白质纳米纤维结构中多样的氨基酸残基使其具有极性和手性,在电场中可操作或用于稳定界面。 [ 75-77 ] 这些结构特征使蛋白质纳米纤维具有很好的材料应用前景,可利用非共价键作用力及共价键作用力自身或复合其它功能材料构筑多维度的、多功能的、生物相容性好的智能材料。或者进一步利用不同的驱动力,构筑具有取向结构的蛋白质纳米纤维材料。
1. 3 基于蛋白质纳米纤维的功能材料
1. 3 .1 一维功能材料
1.3.1.1 蛋白质纳米纤维一维功能材料
蛋白质自组装纳米纤维本身作为一维纳米材料可用作组织支架等起到保护、信息传递等多种生理作用。通过改变纤维化条件,可调控蛋白质自组装结构,获得纳米纤维、纳米条带、 [ 78 ] 纳米管 [ 79 , 80 ] 等不同形貌的一维纳米材料。例如,牙釉蛋白在钙离子及磷酸根存在时,通过调节 pH 4.5-5.5 可得富含 β ‑ 折叠结构的纳米条带,并可作为模板参与牙釉质中磷灰石的生物矿化; [ 78 ] 一种胞嘧啶碱基修饰的多肽( HHQALVFFA )在 pH 3-4 时,胞嘧啶部分质子化,促使多肽自组装形成直径约 24.8 nm 、壁厚约 3.3 nm 的纳米管结构。 [ 81 ] 以水稀释苯丙氨酸二肽的六氟异丙醇溶液,可制备纳米管结构,于管内还原银离子,借助酶降解多肽纳米管,可制备直径约 20 nm 的银纳米线。 [ 82 ] SOD1 基因突变的蛋白质在溶剂挥发的过程中,相邻突变蛋白质 β - 折叠结构间的相互作用可导致纳米管的形成。 [ 83 ]
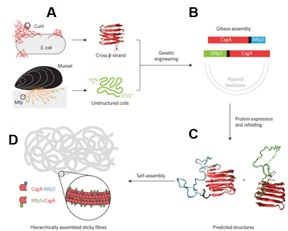
蛋白质纳米纤维作为生物材料,其多肽链中氨基酸序列由转录基因决定。随着基因工程的兴起及其相关技术的发展成熟,通过改变蛋白质基因序列亦可得到具有特定功能的一维纳米材料。例如, Lu 等人通过融合贻贝足蛋白和大肠杆菌黏性淀粉样蛋白序列,成功制备出高粘性的纤维材 料。如 图 1.9 所示,该策略最大限度地结合了贻贝足蛋白和 大肠杆菌黏性淀粉样蛋白黏性特征,所转 录的蛋白质自组装形成纳米纤维并 展现出优良的黏附特性, 其黏附能可达 20 mJ/m2 。 [ 84 ]
图 1. 9 具有水下黏附特性的蛋白质淀粉样纳米纤维的形成步骤。( A )大肠杆菌菌丝中的淀粉样蛋白基因序列与贻贝足蛋白基因序列融合( B ),形成可淀粉样变性的蛋白质基因序列( C ),自组装形成具有粘性的淀粉样变性纤维( D [ 84 ]
1.3.1.2 蛋白质纳米纤维一维复合功能材料
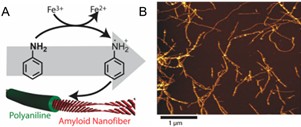
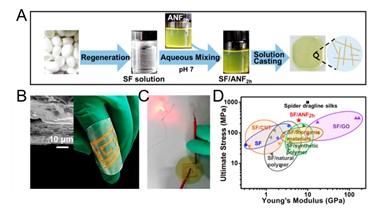
此外,借助蛋白质本身的高反应活性,蛋白质纳米纤维亦常 复合其它纳米材料(如导电高分子、金属及金属氧化物纳米颗粒、碳纳米材料等)获得功能化的一维复合材料。例如, Zhang 等利用蛋白质淀粉样纤维作为模板时利于控制晶面取向及优先暴露低能量晶面的优势,合成了直径为 1.8 nm 的晶态超细铂纳米线,并展现出优异的电催化活性; [ 85 ] Christoph Meier 等利用溶菌酶纳米纤维表面的负电性,通过静电吸附结合正电性的苯胺单体并引发聚合,成功制备出具有核- 壳结构的导电纳米纤维材料。该纤维材料中溶菌酶纤维作为“核”起到模板作用,聚苯胺作为“壳”赋予材料电化学性能,使其可应用于电化学传感、组织工程及电刺激干细胞分化等领域(图 1.10 )。 [ 86 ] Li 等则利用蛋白质与碳纳米管间出色的作用力(包括静电力、疏水力、氢键等), [ 87 ] 使 β - 乳球蛋白在碳纳米管表面纤维化,成功制备出蛋白质自组装纳米纤维包覆的碳纳米管材料。蛋白质纳米纤维包覆层厚度可调,通过近红外光照射或酶处理等可实现材料生物毒性的调节。 [ 88 ]
图 1. 10 ( A )苯胺于蛋白质纳米纤维表面聚合过程示意图。( B )蛋白质纳米纤维 - 聚苯胺复合物原子力显微镜照片。
Figure 1.10 (A) Proposed synthetic pathway of the polymerization of aniline in the vicinity of the amyloid nanofibers. (B) AFM topography images of the PNF−PAni hybrids. [ 86 ]
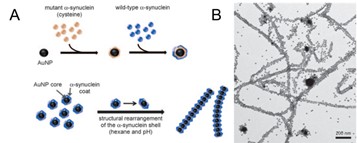
借 助蛋白质的自组装过程,遵循“自下而上”的策略同样可复合金属纳米颗粒等制备一维复合功能材料。例如, Daekyun Lee 等利用 α - 突触核蛋白包覆金纳米颗粒,利用蛋白质纤维化自组装过程,成功实现一维各向异性金纳米颗粒 / 蛋白质纳米纤维复合材料的制备(图 1.11 )。金纳米颗粒在蛋白质纳米纤维上的取向排列使其对普通强度的可见光即可展现很好的光响应性质。 [ 89 ] Sreenath Bolisetty 等人则将 β - 乳球蛋白与磁性氧化铁纳米颗粒混合,并于 pH=3 、 90 °C 下使蛋白质纤维化,得到具有磁性的一维纳米复合材料。 [ 90 ]
图 1. 11 ( A ) α - 突触核蛋白(含有半胱氨酸的突变形式和野生形式)包覆金纳米颗粒(上)及金纳米颗粒在纳米纤维中的取向排列(下)。( B ) pH 诱导所成金纳米颗粒长链的透射电镜图 [ 89 ]
1. 3 .2 二维功能材料
1.3.2.1 蛋白质纳米纤维膜 / 片构筑方法
相对于一维纳米材料,蛋白质纳米纤维作为结构基元构筑二维材料(如片、膜等)操作较为简单,在实际应用中更为广泛。蛋白质纳米纤维长径比高,通过自组装或交联可获得纳米片,例如,一种基于苯丙氨酸二肽的多肽通过芴基保护,在多肽浓度较低时可自组装形成纳米纤维,当多肽浓度升高时,纳米纤维可进一步自组装形成纳米片。 [ 91 ] 而通过简单的自然晾干、真空抽滤、旋涂等方式即可得到稳定、多孔、生物相容性良好的膜材料。例如, β - 乳球蛋白自组装纳米纤维通过真空抽滤,可层层堆叠并形成多孔的膜材料; [ 92 ] 牛胰岛素自组装纳米纤维与聚乙烯醇溶液自然晾干可得强度增大的膜材料; [ 93 ] 修饰了电致发光共轭高分子的胰岛素纳米纤维分散可旋涂制备膜材料,并展现出接近 10 倍的量子效应增强。 [ 94 ]
图 1. 12 纳米膜形成机理示意图。插图展示可在水面自由悬浮的4 英寸纳米膜 [ 95 ]
利用蛋白质分子的两亲性, 当存在两相界面时(固/ 液、液/ 液、气/ 液等),蛋白质材料倾向于在界面处富集,当其空间构型发生变化时可自组装形成纳米纤维结构, [ 96-98 ] 进一步通过氢键及疏水力等分子间作用力可相互交联形成膜材料。 [ 99 ] 例如, Kanishka Biswas 等人将苯丙氨酸二肽溶于水并加入甲苯构成水/ 甲苯两相界面,苯丙氨酸二肽在界面处自组装形成纳米纤维,并进一步组装形成膜材料。利用该界面同样可制备牛胰岛素纳米纤维膜材料。 [ 100 ] Yang 等人从溶菌酶出发,利用还原剂三( 2 - 羧乙基) 膦( TCEP )打开蛋白质分子中的二硫键。该过程诱导溶菌酶分子发生多相成核和自组装过程,自组装溶菌酶蛋白容易在两相界面富集进而形成膜材料。当富集发生在气/ 液界面时可得透明、生物相容性好、厚度可调的膜;当富集发生在固- 液界面时,则可在固体材料表面形成均一、稳定的膜涂层(图 1.12 )。 [ 95 ]
1.3.2.2 蛋白质纳米纤维膜 / 片应用
由蛋白质纳米纤维形成的膜材料具有多孔结构、比表面积大、表面化学基团丰富( -SH 、 -COOH 、 -OH 、 -NH2 等可作为离子结合位点或通过静电吸引结合相反电性的有机染料等 ),且生物相容性高、可生物降解,在污水处理、离子回收、生物探针、组织工程等领域展现出巨大的应用潜力。 例如, Ling 等使用强极性溶剂(六氟异丙醇)剥离桑蚕丝纤维制备蚕丝蛋白纳米纤维,并通过真空抽滤制备透明的自支持膜。该膜厚度最小可达 40 nm ,孔径分布均匀,约为 8-12 nm ,可有效截留有机染料、离子、蛋白质、纳米颗粒等物质。 [ 101 ] 相较于溶液铺膜或再生蚕丝纳米纤维膜,该膜呈现出 更高的透光率。 Tan 等人则在尿素 / 盐酸胍体系中剥离蚕丝蛋白制备蚕丝纳米纤维,通过真空抽滤即可得多孔、溶剂耐受性好、力学强度大的膜材料。除了用于离子、有机染料分子的吸附分离,该膜还可作为固态电池隔膜,使电池展现出优异的电容性能。 [ 102 ]
图 1. 13 多级自组装制备纳米纤维膜。( A )蛋白质分子自组装形成纳米纤维,进而相互堆叠成膜。( B )含塑化剂的溶菌酶纳米纤维膜的偏光显微镜照片 [ 103 ]
另外,蛋白质自组装纳米纤维在体系中趋向于形成热力学向列液晶相,这得益于蛋白质自组装纳米纤维分子结构中的 β - 折叠结构。 β - 折叠结构使蛋白质自组装纳米纤维结构规整,通过分子间相互作用力易于相互堆叠形成取向的结构,可依此构筑具有取向结构的膜材料。例如, Tuomas P. J. Knowles 等人就利用塑化材料表面塑化分子的液晶排列作用,将溶菌酶纳米纤维溶液在其表面铺膜,得到自支撑的、取向排列的溶菌酶纳米纤维薄膜。 [ 103 ] 溶菌酶纳米纤维薄膜力学性能优异,微观尺度下纳米纤维呈现取向排列,展现出明显的偏光现象,结合荧光分子亦可使其取向排列(图 1.13 )。蛋白质纳米纤维取向结构的形成使纤维的膜材料具有特殊的光学特性、力学特性,在细胞培养、生物探针等领域应用广泛。但是,蛋白质纳米纤维的规则排列往往需要精密的实验操作和流程控制,是当下蛋白质纳米纤维膜材料研究的难点和热点。
1.3.2.3 蛋白质纳米纤维二维复合材料
在蛋白质纳米纤维与其他物质形成的复合二维材料方面, Raffaele Mezzenga 等人利用真空抽滤得到 β - 乳球蛋白纳米纤维 / 活性炭复合膜材料。该复合膜材料可有效分离废水中的重金属离子和放射性物质。利用 β - 乳球蛋白纳米纤维的还原性,可将重金属离子加热还原、回收并得到无生物毒性的功能复合膜材料。 [ 104 ] Li 等将石墨烯与 β - 乳球蛋白纳米纤维复合,制备出可降解的纳米复合膜材料。 [ 49 ] 通过简单的真空抽滤过程, β - 乳球蛋白纳米纤维借助疏水力与石墨烯片层结合,可得到多功能的膜材料。该膜材料中, β - 乳球蛋白纳米纤维可被酶降解从而引起材料导电性的变化,以此可实现对酶的检测。同时,该膜材料具有形状记忆功能,可用于湿度检测等(图 1.14 )。 Li 等进一步以蛋白质纳米纤维复合羟基磷灰石纳米片,成功制备出类骨骼结构的仿生纳米复合膜材料。 [ 105 ] 蛋白质纳米纤维 / 羟基磷灰石纳米片仿生膜材料具有典型的珍珠层结构,力学性能优异,对成骨细胞的吸附和生长增殖可媲美商用基底。 Sreenath Bolisetty 等人以金 / 钯纳米颗粒修饰蛋白质纳米纤维,构筑了可用于流动催化的复合膜材料。该膜可有效催化 4- 硝基苯酚的还原反应,纳米纤维的高长径比及与金 / 钯纳米颗粒良好的结合力,在流动催化时可有效防止催化组分的损失,维持催化活性的持久高效。 [ 92 ]
图 1. 14 β - 乳球蛋白纳米纤维与石墨烯复合示意图。( A ) β - 乳球蛋白于 pH 2 、 90 °C 自组装形成纳米纤维。( B )蛋白质纳米纤维与氧化石墨烯通过静电作用结合。( C )氧化石墨烯于 80 °C 剧烈搅拌还原,断裂的蛋白质纳米纤维优先在其表面吸附。( D )真空抽滤制备蛋白质纳米纤维/ 石墨烯纳米片复合膜 [ 49 ]
蛋白质自组装纳米纤维分子结构中的 β - 折叠结构一方面赋予膜材料出色的力学性能,另一方面也导致膜材料刚性大,限制材料的可操作性。很多研究期望通过引入第二组分,借助于第二组分特性或其与蛋白质纳米纤维的相互作用提高膜材料的力学强度。合成高分子作为长链聚合物,借由氢键、静电吸附等非共价作用力或化学交联可与蛋白质纳米纤维复合形成强度提升的膜材料。蛋白质纳米纤维 / 合成高分子复合膜材料同时具备高力学强度、柔韧性以及生物相容性等特点,常用于柔性电极、可穿戴智能设备等领域。例如, Lv 等人将自然界中强度较大的再生蚕丝纳米纤维与合成高分子中强度较大的芳纶纳米纤维混合,通过简单的溶液铺膜过程制备出力学性能优异、生物相容性佳的复合膜材料(图 1.15 ); [ 47 ] 优良的力学性能使该复合膜材料可用于高压过滤分离过程。 Gerrard 等人将牛胰岛素自组装纳米纤维与高分子聚乙烯醇复合,通过溶剂蒸发制备具有良好力学强度的膜材料。该膜杨氏模量大,但复合过程导致高分子微观自由体积减小,玻璃化转变温度降低。研究者指出,利用蛋白质纳米纤维的表面化学改性可能实现复合材料热力学性能的改善。 [ 93 ] 除了引入第二组分,从蛋白质纳米纤维结构调控或膜材料制备方法入手提高材料的柔韧性,可最大限度保持蛋白质纳米纤维的材料及物化特性,但过程相对复杂,需要更多系统的研究。
图 1. 15 ( A )三步法制备再生蚕丝纳米纤维/芳纶纳米纤维复合膜。即再生蚕丝纳米纤维制备、水溶液混合芳纶纳米纤维和溶液铺膜。( B )金微米片在复合膜上沉积形成柔性环形电路。插图展示金微米片在膜上的沉积。( C )柔性环形电路导电性评价。( D )蚕丝蛋白/芳纶纳米纤维复合膜与其它基于蚕丝蛋白材料的力学性能比较 [ 47 ]
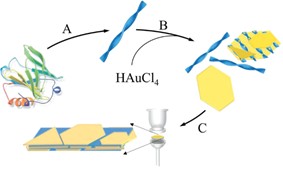
除直接复合形成二维功能材料外,蛋白质纳米纤维还可作为还原剂、稳定剂或封端剂参与纳米材料的制备过程,进而获得功能化二维材料。例如, Li 等人利用 β - 乳球蛋白纳米纤维作为还原剂和封端剂,辅助氯金酸还原,得到表面粘附了 β - 乳球蛋白纳米纤维的二维金片,通过真空抽滤制得功能复合膜材料(图 1.16 ) ,展现出良好的湿度响应,并很好的保持了材料的荧光特性。 [ 6 ] 蛋白质纳米纤维一方面用于还原制备金片并形成稳定的胶体溶液,另一方面其高强度、高长径比特性亦有助于承受真空抽滤过程,可有效避免过程中有机组分的损失。 Samit K. Ray 等人将丝素蛋白的还原能力和自组装性能相结合,通过陈化丝素蛋白与 HAuCl4 的混合溶液,还原形成金纳米颗粒,同时丝素蛋白分子自组装形成纳米纤维。而后通过浇铸成膜的方法在铂电极上形成精密复杂的图案,展现出良好的光电检测性能。 [ 106 ] Buehler 等人 利用六氟异丙醇剥离蚕丝蛋白并超声分散得到蚕丝纳米纤维分散液,借助蚕丝蛋白的还原能力,还原氯金酸盐制备金纳米片,通过真空抽滤构筑了蚕丝蛋白纳米纤维/ 金纳米片可穿戴电极。 [ 107 ] 除还原金属离子外,蛋白质纳米纤维还可参与羟基磷灰石的矿化过程,所得复合材料可用于组织工程。 [ 108 ] Hakan Ceylan 等人即利用多肽自组装纳米纤维促进矿化过程进行,制备多肽纳米纤维/ 磷酸钙纳米颗粒复合膜,可用于人骨髓干细胞培养,促进骨修复。 [ 109 ]
图 1. 16 纳米复合材料制备示意图。( A ) β - 乳球蛋白自组装形成纳米纤维; ( B )蛋白质纳米纤维还原制备并稳定金片; ( C )真空抽滤法制备 β - 乳球蛋白纳米纤维- 金片复合材料 [ 6 ]
1. 3 .3 三维功能材料
1.3.3.1 蛋白质纳米纤维三维材料构筑
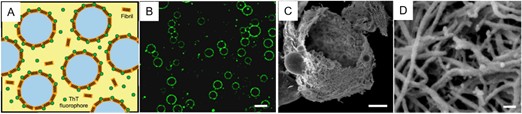
蛋白质纳米纤维作为结构单元可构筑结构复杂、生物相容性好的功能材料,如水凝胶、气凝胶、微乳液、取向材料等,并在组织支架、药物及活性分子载体等领域有出色应用。 [ 110 , 111 ] 基于蛋白质纳米纤维出色的化学活性和比表面积等特性,通过改变体系温度、 pH 、离子强度或者加入交联剂(如戊二醛、丁烷四羧酸)等,可使蛋白质纳米纤维彼此交联形成微观具有网状结构的三维功能材料。 [ 112 ] 通常,蛋白质纳米纤维通过物理交联或化学交联的方法实现三维功能材料的构筑。物理交联是指借助于蛋白质纳米纤维的结构和化学特性,通过静电吸引、疏水力、氢键、范德华力、 π-π 相互作用等非共价作用力实现的交联。 [ 113 , 114 ] 例如, Reeba S. Jacob 等人通过制备 Aβ 42 自组装纳米纤维并借助于纤维间的疏水力、氢键及 π-π 相互作用成功构筑了具有自愈合性质的水凝胶。该凝胶具有出色的生物相容性,可用于多种细胞的吸附和培养。 [ 8 ] Marta J. Krysmann 等成功制备了 Aβ (16–20) 自组装纳米纤维,并通过提高缓冲液浓度,使纳米纤维间静电排斥力减弱,构筑了蛋白质纳米纤维水凝胶。 [ 115 ]
不同于物理交联,化学交联利用蛋白质纳米纤维氨基酸残基的多样性( NH2 、 OH 、 COOH 、 SH 等),辅助以交联剂分子(戊二醛、丁烷四羧酸等),通过化学反应使蛋白质纳米纤维彼此交联形成三维功能材料。例如, Joel H. Collier 等人利用加热或近红外光照射的方式,诱导形成多肽自组装纳米纤维水凝胶。这种刺激响应的特性,可以使其应用在药物释放和伤口愈合等领域。 [ 116 ] Raffaele Mezzenga 借助于交联剂丁烷四羧酸与蛋白质纳米纤维羟基的交联反应,成功制备出基于 β - 乳球蛋白纳米纤维的水凝胶。通过冻干过程,可以对所得水凝胶的孔径大小进行调控,并可很好地应用于细胞培养。 [ 117 ]
1.3.3.2 蛋白质纳米纤维三维复合材料
蛋白质纳米纤维与其他材料杂化形成三维复合功能性材料,亦分为直接复合和化学交联复合两种方式。例如, Raffaele Mezzenga 等人采用 β - 乳球蛋白纳米纤维为模板,复合金纳米颗粒或金纳米片,借助于离子交联和超临界二氧化碳干燥,成功制备出蛋白质纳米纤维复合气凝胶。如图 1.17 所示,该气凝胶孔隙率高,且可自发荧光,借助于金纳米组分的特性,亦可用于压力传感和催化领域。 [ 118 ] Tang 等人利用高温诱导牛血清白蛋白自组装形成纳米纤维网状结构,进而原位聚合功能高分子得到具有双网络结构的复合凝胶材料。与传统蛋白质凝胶相比,该凝胶展现出优良的力学性能(杨氏模量: 252−1199 kPa 、强度: 0.24−0.48 MPa 、断裂能: 3.56−16.88 MJ/m3 、拉伸性能: 7.7−79.9 mm/mm )、快速恢复以及出色的粘附性能。 [ 119 ] Li 等则利用聚( N- 异丙基丙烯酰胺)( PNiPAM )包覆 β - 乳球蛋白纳米纤维 ,使其在低于临界凝胶浓度时亦可形成稳定凝胶材料,展现出温度响应特性。 [ 120 ]
图 1. 17 蛋白质纳米纤维气凝胶及金 / 蛋白质纳米纤维气凝胶照片及扫描电镜照片。 (A-C) 蛋白质纳米纤维、金纳米颗粒 / 蛋白质纳米纤维、金片 / 蛋白质纳米纤维气凝胶照片。( D-F )蛋白质纳米纤维、金纳米颗粒 / 蛋白质纳米纤维、金片 / 蛋白质纳米纤维气凝胶扫描电镜照片[ 118 ]
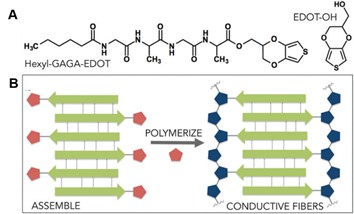
1. 18 ( A )己基 -GAGA-EDOT 和 EDOT-OH 结构式。( B )自组装结构及其聚合过程示意图 [ 121 ]
通过化学交联同样可制备蛋白质纳米纤维复合凝胶材料。例如, Ken-Ichi Sano 等人利用微管蛋白与聚乙二醇的交联反应,得到温度响应的水凝胶。通过微管蛋白组装过程的可逆性,在温度范围 4-37 °C 内可使凝胶进行溶胶 - 凝胶转化。 [ 122 ] T. J. Blatz 等人则从分子设计角度出发,首先将噻吩基团化学接枝到小分子四肽上,利用四肽分子的自组装性能形成纳米纤维,进而利用噻吩分子的聚合性能,得到导电的水凝胶材料(图 1.18 )。该凝胶导电率可达 10-3 S/cm ,可作为刺激响应的仿生智能材料。 [ 121 ]
1.3.3.3 蛋白质纳米纤维取向结构
图 1. 19 (A) 凝胶线双折射结果,表明多肽纳米纤维沿凝胶线轴向的取向排列。( B )取向排列多肽纳米纤维的扫描电镜图片 [ 125 ]
蛋白质自组装纳米纤维在其三维材料中多为无规排列,借助不同的驱动力,使蛋白质纳米纤维取向排列并逐级组装还可形成三维尺度的、具有各向异性的材料,有利于材料力学性能优化,光学性质调控及仿生材料构筑等。对于蛋白质纳米纤维取向材料的构筑,可借助于外部场力(如电场、磁场等)、剪切力、空间约束以及蛋白质分子的自组装性能。 例如,通过几何或者力学约束,蛋白质分子在特定的空间或各向异性力的作用下自组装形成纳米纤维,并进一步规则排列得到长程有序的取向结构。 [ 123 ] 借助于注射器产生的剪切力,一维多肽纳米纤维沿作用力方向运动并通过氢键、静电吸引等分子间作用力相互交联,得到宏观尺寸为厘米级别的具有取向结构的多肽纳米纤维凝胶线(图 1.19 )。 [ 124 , 125 ] 与此类似,通过流动聚集同样可以调控蛋白质纳米纤维自组装形成具有取向结构的微米级纤维材料。 [ 126 ]
然而,蛋白质纳米纤维在三维尺度上的规则排列仍然较难实现,外部场力的高耗能及蛋白质自组装过程的复杂性等是取向材料构筑方法的缺陷。因此,寻找新的蛋白质纳米纤维取向材料制备方法,在低能耗、操作简单、易于控制等优势下使纳米纤维定向排列是亟待解决的难题。
1.3.3.4 蛋白质纳米纤维稳定乳液
除 具有精密结构的取向材料外,借助蛋白质纳米纤维的两亲性,其在两相界面的聚集和组装亦可促进微乳液的形成并起到良好的稳定作用。蛋白质纳米纤维稳定的微乳液具有良好的生物相容性,在细胞支架及功能囊等领域展现了巨大的应用前景。 [ 127 ] 例如, Ulyana Shimanovich 等人利用微流控技术,以溶菌酶纳米纤维稳定油 / 水微乳液,得到均一分散的微乳液体系。该微乳液可作为药物载体(如盘尼西林等)做到可控释放、并且展现出优良的生物相容性。 [ 111 ] Zhou 等人首先利用微流控技术将酶分子包覆在微乳液中,进而使表层酶分子纤维化,得到稳定的微乳液。该微乳液体系保持了酶的活性并可实现酶的可控释放。 [ 128 ] 除了常规的油 - 水界面,蛋白质纳米纤维同样可在全水相体系中形成稳定的乳液。如图 1.20 所示, Song 等人将互不相溶的葡萄聚糖和聚乙二醇溶液混合,并以溶菌酶纳米纤维作为稳定剂,成功制备出全水相的乳液体系。 [ 97 ] 这种全水相的乳液体系相对于传统的油 / 水乳液体系生物相容性更高,可用做药物载体或细胞培养基质等。
图 1. 20 溶菌酶纳米纤维在水 / 水乳液界面富集( A )示意图及( B ) ThT 染色荧光图,标尺: 20 毫米。( C,D )多层溶菌酶纳米纤维所构成囊泡的扫描电镜图片,标尺: 2 微米( C )、 50 纳米( D ) [ 97 ]
参考文献
[1] LIAO Y, CHANG Y, YOSHIIKE Y, et al . Negatively charged gold nanoparticles inhibit Alzheimer's amyloid-β fibrillization, induce fibril dissociation, and mitigate neurotoxicity [J]. Small, 2012, 8 (23): 3631-3639.
[2] HARPER J D, PETER T, LANSBURY J. Models of amyloid seeding in Alzheimer's disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins [J]. Annual Review of Biochemistry, 1997, 66 (1): 385-407.
[3] ROYCHAUDHURI R, YANG M, Hoshi M M, et al. Amyloid β-protein assembly and Alzheimer disease [J]. Journal of Biological Chemistry, 2009, 284 (8): 4749-4753.
[4] Dobson C M. Protein folding and misfolding. Nature, 2003, 426 (6968): 884-890.
[5] Tang Q, Solin N, Lu J, et al. Hybrid bioinorganic insulin amyloid fibrils [J]. Chemical Communications, 2010, 46 (23): 4157-4159.
[6] Li C, Bolisetty S, Mezzenga R. Hybrid nanocomposites of gold single-crystal platelets and amyloid fibrils with tunable fluorescence, conductivity, and sensing properties [J]. Advanced Materials, 2013, 25 (27): 3694-3700.
[7] Li C, Mezzenga R. Functionalization of multiwalled carbon nanotubes and their pH-responsive hydrogels with amyloid fibrils [J]. Langmuir, 2012, 28 (27): 10142-10146.
[8] Jacob R S, Ghosh D, Singh P K, et al. Self healing hydrogels composed of amyloid nano fibrils for cell culture and stem cell differentiation [J]. Biomaterials, 2015, 54: 97-105.
[9] Mains J, Lamprou D A, McIntosh L, et al. Beta-adrenoceptor antagonists affect amyloid nanostructure; amyloid hydrogels as drug delivery vehicles [J]. Chemical Communications, 2013, 49 (44): 5082-5084.
[10] Fandrich M. On the structural definition of amyloid fibrils and other polypeptide aggregates [J]. Cellular and Molecular Life Sciences, 2007, 64 (16): 2066-2078.
[11] Cao B, Zhao Y, Kou Y, et al. Structure of the nonameric bacterial amyloid secretion channel [J]. Proceedings of the National Academy of Sciences of the United States of America, 2014, 111 (50): 5439-5444.
[12] Irvine G B, El-Agnaf O M, Shankar G M, et al. Protein aggregation in the brain: The molecular basis for Alzheimer's and Parkinson's diseases [J]. Molecular Medicine, 2008, 14 (7-8): 451-464.
[13] Verma M, Vats A, Taneja V. Toxic species in amyloid disorders: Oligomers or mature fibrils [J]. Annals of Indian Academy of Neurology, 2015, 18 (2): 138-145.
[14] Paravastu A K, Leapman R D, Yau W -M, et al. Molecular structural basis for polymorphism in Alzheimer's beta-amyloid fibrils [J]. Proceedings of the National Academy of Sciences of the United States of America, 2008, 105 (47): 18349-18354.
[15] Lambert M P, Barlow A K, Chromy B A, et al. Diffusible, nonfibrillar ligands derived from A beta(1-42) are potent central nervous system neurotoxins [J]. Proceedings of the National Academy of Sciences of the United States of America, 1998, 95 (11): 6448-6453.
[16] Hammarstrom. P, Wiseman R L, Powers E T, et al. Prevention of transthyretin amyloid disease by changing protein misfolding energetics [J]. Science, 2003, 299 (5607): 713-716.
[17] Das P, Kang S -g, Temple S, et al. Interaction of amyloid inhibitor proteins with amyloid beta peptides: Insight from molecular dynamics simulations [J]. Plos One, 2014, 9 (11): 0113041.
[18] Porat Y, Abramowitz A, Gazit E. Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism [J]. Chemical Biology & Drug Design, 2006, 67 (1): 27-37.
[19] Reinke A A, Gestwicki J E. Structure-activity relationships of amyloid beta-aggregation inhibitors based on curcumin: Influence of linker length and flexibility [J]. Chemical Biology & Drug Design, 2007, 70 (3): 206-215.
[20] Scarmeas N, Stern Y, Tang M X, et al. Mediterranean diet and risk for Alzheimer's disease [J]. Annals of Neurology, 2006, 59 (6): 912-921.
[21] Sashina E S, Bochek A M, Novoselov N P, et al. Structure and solubility of natural silk fibroin [J]. Russian Journal of Applied Chemistry, 2006, 79 (6): 869-876.
[22] Cheng C, Teasdale I, Bruggemann O. Stimuli-responsive capsules prepared from regenerated silk fibroin microspheres [J]. Macromolecular Bioscience, 2014, 14 (6): 807-816.
[23] Zeng L K, Jiang L N, Teng W B, et al. Engineering aqueous fiber assembly into silk-elastin-like protein polymers [J]. Macromolecular Rapid Communications, 2014, 35 (14): 1273-1279.
[24] Chan H M, Li H W. Multifunctional encoded self-assembling protein nanofibrils as platform for high-throughput and multiplexed detection of biomolecules [J]. Analytical Chemistry, 2011, 83 (24): 9370-9377.
[25] Knowles T P J, Mezzenga R . Amyloid fibrils as building blocks for natural and artificial functional materials [J]. Advanced Materials, 2016, 28 (31): 6546-6561.
[26] Kasai S, Ohga Y, Mochizuki M, et al. Multifunctional peptide fibrils for biomedical materials [J]. Biopolymers, 2004, 76 (1): 27-33.
[27] Sasso L, Suei S, Domigan L, et al. Versatile multi-functionalization of protein nanofibrils for biosensor applications [J]. Nanoscale, 2014, 6 (3): 1629-1634.
[28] Fowler D M, Koulov A V, Balch W E, et al. Functional amyloid - from bacteria to humans [J]. Trends in Biochemical Sciences, 2007, 32 (5): 217-224.
[29] Krishnan R, Lindquist S L. Structural insights into a yeast prion illuminate nucleation and strain diversity [J]. Nature, 2005, 435 (7043): 765-772.
[30] Tanaka M, Collins S R, Toyama B H, et al. The physical basis of how prion conformations determine strain phenotypes [J]. Nature, 2006, 442 (7102): 585-589.
[31] Collinson S K, Doig P C, Doran J L, et al. Thin, aggregative fimbriae mediate binding of salmonella-enteritidis to fibronectin [J]. Journal of Bacteriology, 1993, 175 (1): 12-18.
[32] Gebbink M, Claessen D, Bouma B, et al. Amyloids - A functional coat for microorganisms [J]. Nature Reviews Microbiology, 2005, 3 (4): 333-341.
[33] Zeng G, Vad B S, Dueholm M S, et al. Functional bacterial amyloid increases pseudomonas biofilm hydrophobicity and stiffness [J]. Frontiers in Microbiology, 2015, 6: 1-14.
[34] Schwartz K, Syed A K, Stephenson R E, et al. Functional amyloids composed of phenol soluble modulins stabilize staphylococcus aureus biofilms [J]. Plos Pathogens, 2012, 8 (6): 1-11.
[35] Dueholm M S, Petersen S V, Sonderkaer M, et al. Functional amyloid in pseudomonas [J]. Molecular Microbiology, 2010, 77 (4): 1009-1020.
[36] Iconomidou V A, Vriend G, Hamodrakas S J. Amyloids protect the silkmoth oocyte and embryo [J]. Febs Letters, 2000, 479 (3): 141-145.
[37] Jordens S, Isa L, Usov I, et al. Non-equilibrium nature of two-dimensional isotropic and nematic coexistence in amyloid fibrils at liquid interfaces [J]. Nature Communications, 2013, 4: 1-8.
[38] Jung J -M, Gunes D Z, Mezzenga R . Interfacial activity and interfacial shear rheology of native beta-lactoglobulin monomers and their heat-induced fibers [J]. Langmuir, 2010, 26 (19): 15366-15375.
[39] Ruehs P A, Scheuble N, Windhab E J, et al. Protein adsorption and interfacial rheology interfering in dilatational experiment [J]. European Physical Journal-Special Topics, 2013, 222 (1): 47-60.
[40] Mostaert A S, Higgins M J, Fukuma T , et al. Nanoscale mechanical characterisation of amyloid fibrils discovered in a natural adhesive [J]. Journal of Biological Physics, 2006, 32 (5): 393-401.
[41] Fowler D M, Koulov A V, Alory-Jost C, et al. Functional amyloid formation within mammalian tissue [J]. Plos Biology, 2006, 4 (1): 100-107.
[42] Maji S K, Perrin M H, Sawaya M R, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules [J]. Science, 2009, 325 (5938): 328-332.
[43] An B, Wu X, Li M, et al. Hydrophobicity-modulating self-assembled morphologies of α-Zein in aqueous ethanol [J]. International Journal of Food Science & Technology, 2016, 51 (12): 2621-2629.
[44] Reddy N, Yang Y. Novel Protein fibers from wheat gluten [J]. Biomacromolecules, 2007, 8 (2): 638-643.
[45] Juárez J, Cambón A, Goy-López S, et al. Obtention of metallic nanowires by protein biotemplating and their catalytic application [J]. Journal of Physical Chemistry Letters, 2010, 1 (18): 2680-2687.
[46] Li C, Adamcik J, Mezzenga R. Biodegradable nanocomposites of amyloid fibrils and graphene with shape-memory and enzyme-sensing properties [J]. Nature Nanotechnology, 2012, 7 (7): 421-427.
[47] Lv L, Han X, Zong L, et al. Biomimetic hybridization of kevlar into silk fibroin: nanofibrous strategy for improved mechanic properties of flexible composites and filtration membranes [J]. Acs Nano, 2017, 11 (8): 8178-8184.
[48] Meisl G, Yang X, Hellstrand E, et al. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 Peptides [J]. Proceedings of the National Academy of Sciences of the United States of America, 2014, 111 (26): 9384-9389.
[49] Li C, Adamcik J, Mezzenga R. Biodegradable nanocomposites of amyloid fibrils and graphene with shape-memory and enzyme-sensing properties [J]. Nature Nanotechnology, 2012, 7 (7): 421-427.
[50] Wu X, Han X, Lv L, et al. Supramolecular proteinaceous biofilms as trapping sponges for biologic water treatment and durable catalysis [J]. Journal of Colloid and Interface Science, 2018, 527: 117-123.
[51] Wu X, Li M, Li Z, et al. Amyloid-graphene oxide as immobilization platform of Au nanocatalysts and enzymes for improved glucose-sensing activity [J]. Journal of Colloid and Interface Science, 2017, 490: 336-342.
[52] Mankar S, Anoop A, Sen S, et al. Nanomaterials: amyloids reflect their brighter side [J]. Nano reviews, 2011, 2 : 6032.
[53] Li C, Mezzenga R . The interplay between carbon nanomaterials and amyloid fibrils in bio-nanotechnology [J]. Nanoscale, 2013, 5 (14): 6207-6218.
[54] Yoo S I, Yang M, Brender J R, et al. Inhibition of amyloid peptide fibrillation by inorganic nanoparticles: functional similarities with proteins [J]. Angewandte Chemie-International Edition, 2011, 50 (22): 5110-5115.
[55] Cabaleiro-Lago C, Quinlan-Pluck F, Lynch I, et al. Dual Effect of amino modified polystyrene nanoparticles on amyloid beta protein fibrillation [J]. Acs Chemical Neuroscience, 2010, 1 (4): 279-287.
[56] Takahashi T, Mihara H. Peptide and protein mimetics inhibiting amyloid beta-peptide aggregation [J]. Accounts of Chemical Research, 2008, 41 (10): 1309-1318.
[57] Milojevic J, Esposito V, Das R, et al. Understanding the molecular basis for the inhibition of the Alzheimer's A beta-peptide oligomerization by human serum albumin using saturation transfer difference and off-resonance relaxation NMR spectroscopy [J]. Journal of the American Chemical Society, 2007, 129 (14): 4282-4290.
[58] Linse S, Cabaleiro-Lago C, Xue W -F, et al. Nucleation of protein fibrillation by nanoparticles [J]. Proceedings of the National Academy of Sciences of the United States of America, 2007, 104 (21): 8691-8696.
[59] Grigolato F, Colonabo C, Ferrari R, et al. Mechanistic origin of the combined effect of surfaces and mechanical agitation on amyloid formation [J]. Acs Nano, 2017, 11 (11): 11358-11367.
[60] Campioni S, Carret G, Jordens S, et al. The presence of an air-water interface affects formation and elongation of alpha-synuclein fibrils [J]. Journal of the American Chemical Society, 2014, 136 (7): 2866-2875.
[61] Viles J H . Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer's, Parkinson's and prion diseases [J]. Coordination Chemistry Reviews, 2012, 256 (19-20): 2271-2284.
[62] Calabrese M F, Miranker A D . Metal binding sheds light on mechanisms of amyloid assembly [J]. Prion, 2009, 3 (1): 1-4.
[63] Chen W -T, Liao Y -H, Yu H -M, et al. Distinct effects of Zn2+ , Cu2+ , Fe3+ , and Al3+ on amyloid-beta stability, oligomerization, and aggregation [J]. Journal of Biological Chemistry, 2011, 286 (11): 9646-9656.
[64] Sarell C J, Wilkinson S R, Viles J H. Substoichiometric levels of Cu2+ ions accelerate the kinetics of fiber formation and promote cell toxicity of amyloid-beta from Alzheimer disease [J]. Journal of Biological Chemistry, 2010, 285 (53): 41533-41540.
[65] Fitzpatrick A W P, Debelouchina G T, Bayro M J, et al. Atomic structure and hierarchical assembly of a cross-beta amyloid fibril [J]. Proceedings of the National Academy of Sciences of the United States of America, 2013, 110 (14): 5468-5473.
[66] Wasmer C, Lange A, Van Melckebeke H, et al. Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core [J]. Science, 2008, 319 (5869): 1523-1526.
[67] Rambaran R N, Serpell L C. Amyloid fibrils abnormal protein assembly [J]. Prion, 2008, 2 (3) : 112-117.
[68] Malmos K G, Blancas-Mejia L M, Weber B , et al. ThT 101: a primer on the use of thioflavin T to investigate amyloid formation [J]. Amyloid-Journal of Protein Folding Disorders, 2017, 24 (1): 1-16.
[69] Khan J M, Khan M S, Ali M S , et al. Cetyltrimethylammonium bromide (CTAB) promote amyloid fibril formation in carbohydrate binding protein (concanavalin A) at physiological pH [J]. Rsc Advances, 2016, 6 (44): 38100-38111.
[70] Fandrich M, Fletcher M A, Dobson C M . Amyloid fibrils from muscle myoglobin - Even an ordinary globular protein can assume a rogue guise if conditions are right [J]. Nature, 2001, 410 (6825): 165-166.
[71] Miller L M, Bourassa M W, Smith R J . FTIR spectroscopic imaging of protein aggregation in living cells [J]. Biochimica Et Biophysica Acta-Biomembranes, 2013, 1828 (10): 2339-2346.
[72] Eisenberg D, Jucker M . The amyloid state of proteins in human diseases [J]. Cell, 2012, 148 (6): 1188-1203.
[73] Aymard P, Nicolai T, Durand D , et al. Static and dynamic scattering of beta-lactoglobulin aggregates formed after heat-induced denaturation at pH 2 [J]. Macromolecules, 1999, 32 (8): 2542-2552.
[74] Knowles T P, Fitzpatrick A W, Meehan S,, et al. Role of intermolecular forces in defining material properties of protein nanofibrils [J]. Science, 2007, 318 (5858): 1900-1903.
[75] Rogers S S, Venema P, Van der Ploeg J P M, et al. Investigating the permanent electric dipole moment of beta-lactoglobulin fibrils, using transient electric birefringence [J]. Biopolymers, 2006, 82 (3): 241-252.
[76] Jordens S, Riley E E, Usov I, et al. Adsorption at liquid interfaces induces amyloid fibril bending and ring formation [J]. Acs Nano, 2014, 8 (11): 11071-11079.
[77] Gao G, Zhang M, Lu P, et al. Chirality-assisted ring-like aggregation of a beta(1-40) at liquid-solid interfaces: A stereoselective two-step assembly process [J]. Angewandte Chemie-International Edition, 2015, 54 (7): 2245-2250.
[78] Carneiro K M M, Zhai H, Zhu L , et al. Amyloid-like ribbons of amelogenins in enamel mineralization [J]. Scientific Reports, 2016, 6: 1-11.
[79] Kumara M T, Srividya N, Muralidharan S , et al. Bioengineered flagella protein nanotubes with cysteine loops: Self-assembly and manipulation in an optical trap [J]. Nano Letters, 2006, 6 (9): 2121-2129.
[80] Brodin J D, Smith S J, Carr J R, et al. Designed, helical protein nanotubes with variable diameters from a single building block [J]. Journal of the American Chemical Society, 2015, 137 (33): 10468-10471.
[81] Liu P, Ni R, Mehta A K , et al. Nucleobase-directed amyloid nanotube assembly [J]. Journal of the American Chemical Society, 2008, 130 (50): 16867-16869.
[82] Reches M, Gazit E . Casting metal nanowires within discrete self-assembled peptide nanotubes [J]. Science, 2003, 300 (5619): 625-627.
[83] Elam J S, Taylor A B, Strange R, et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS [J]. Nature Structural Biology, 2003, 10 (6): 461-467.
[84] Zhong C, Gurry T, Cheng A A, et al. Strong underwater adhesives made by self-assembling multi-protein nanofibres [J]. Nature Nanotechnology, 2014, 9 (10): 858-866.
[85] Zhang L, Li N, Gao F, et al. Insulin amyloid fibrils: an excellent platform for controlled synthesis of ultrathin superlong platinum nanowires with high electrocatalytic activity [J]. Journal of the American Chemical Society, 2012, 134 (28): 11326.
[86] Meier C, Lifincev I, Welland M E. Conducting core-shell nanowires by amyloid nanofiber templated polymerization [J]. Biomacromolecules, 2015, 16 (2): 558-563.
[87] Sadana A. Protein adsorption and inactivation on surfaces - Influence of heterogeneities [J]. Chemical Reviews, 1992, 92 (8): 1799-1818.
[88] Li C, Bolisetty S, Chaitanya K, et al. Tunable carbon nanotube/protein core-shell nanoparticles with NIR- and enzymatic-responsive cytotoxicity [J]. Advanced Materials, 2013, 25 (7): 1010-1015.
[89] Lee D, Choe Y -J, Choi Y S, et al. Photoconductivity of pea-pod-type chains of gold nanoparticles encapsulated within dielectric amyloid protein nanofibrils of alpha-synuclein [J]. Angewandte Chemie-International Edition, 2011, 50 (6): 1332-1337.
[90] Bolisetty S, Vallooran J J, Adamcik J, et al. Magnetic-responsive hybrids of Fe3 O4 nanoparticles with beta-lactoglobulin amyloid fibrils and nanoclusters [J]. Acs Nano, 2013, 7 (7): 6146-6155.
[91] Qin S -Y, Pei Y, Liu X -J, et al. Hierarchical self-assembly of a beta-amyloid peptide derivative [J]. Journal of Materials Chemistry B, 2013, 1 (5): 668-675.
[92] Bolisetty S, Arcari M, Adamcik J , et al. Hybrid amyloid membranes for continuous flow catalysis [J]. Langmuir, 2015, 31 (51): 13867-13873.
[93] Rao S P, Meade S J, Healy J P, et al. Amyloid fibrils as functionalizable components of nanocomposite materials [J]. Biotechnology Progress, 2012, 28 (1): 248-256.
[94] Tanaka H, Herland A, Lindgren L J, et al. Enhanced current efficiency from bio-organic light-emitting diodes using decorated amyloid fibrils with conjugated polymer [J]. Nano Letters, 2008, 8 (9): 2858-2861.
[95] Wang D, Ha Y, Gu J , et al. 2D Protein Supramolecular nanofilm with exceptionally large area and emergent functions [J]. Advanced Materials, 2016, 28 (34): 7414-7423.
[96] Ukraintsev E V, Kiselev G A, Kudrinskii A A, et al. Formation of lysozyme fibrils on a solid support [J]. Polymer Science Series B, 2007, 49 (1-2): 6-9.
[97] Song Y, Shimanovich U, Michaels T C T, et al. Fabrication of fibrillosomes from droplets stabilized by protein nanofibrils at all-aqueous interfaces [J]. Nature Communications, 2016, 7: 1-8.
[98] Shen L, Adachi T, Vanden Bout D, et al. A mobile precursor determines amyloid-beta peptide fibril formation at interfaces [J]. Journal of the American Chemical Society, 2012, 134 (34): 14172-14178.
[99] Hendler N, Sidelman N, Reches M, et al. Formation of well-organized self-assembled films from peptide nanotubes [J]. Advanced Materials, 2007, 19 (11): 1485-1488.
[100] Biswas K, Rao C N R. Nanostructured peptide fibrils formed at the organic-aqueous interface and their use as templates to prepare inorganic nanostructures [J]. Acs Applied Materials & Interfaces, 2009, 1 (4): 811-815.
[101] Ling S, Jin K, Kaplan D L, et al. Ultrathin free-standing bombyx mori silk nanofibril membranes [J]. Nano Letters, 2016, 16 (6): 3795-3800.
[102] Tan X X, Zhao W C, Mu T C . Controllable exfoliation of natural silk fibers into nanofibrils by protein denaturant deep eutectic solvent: nanofibrous strategy for multifunctional membranes [J]. Green Chemistry, 2018, 20 (15): 3625-3633.
[103] Knowles T P J, Oppenheim T W, Buell A K , et al. Nanostructured films from hierarchical self-assembly of amyloidogenic proteins [J]. Nature Nanotechnology, 2010, 5 (3): 204-207.
[104] Bolisetty S, Mezzenga R . Amyloid-carbon hybrid membranes for universal water purification [J]. Nature Nanotechnology, 2016, 11 (4): 365-371.
[105] Li C, Born A -K, Schweizer T, et al. Amyloid-hydroxyapatite bone biomimetic composites [J]. Advanced Materials, 2014, 26 (20): 3207-3212.
[106] Gogurla N, Sinha A K, Naskar D, et al. Metal nanoparticles triggered persistent negative photoconductivity in silk protein hydrogels [J]. Nanoscale, 2016, 8 (14): 7695-7703.
[107] Ling S, Li C, Jin K , et al. Liquid exfoliated natural silk nanofibrils: Applications in optical and electrical devices [J]. Advanced Materials, 2016, 28 (35): 7783-7790.
[108] Wei G, Reichert J, Bossert J, et al. Novel biopolymeric template for the nucleation and growth of hydroxyapatite crystals based on self-assembled fibrinogen fibrils [J]. Biomacromolecules, 2008, 9 (11): 3258-3267.
[109] Ceylan H, Kocabey S, Gulsuner H U, et al. Bone-like mineral nucleating peptide nanofibers induce differentiation of human mesenchymal stem cells into mature osteoblasts [J]. Biomacromolecules, 2014, 15 (7): 2407-2418.
[110] Kopecek J, Yang J . Peptide-directed self-assembly of hydrogels [J]. Acta Biomaterialia, 2009, 5 (3): 805-816.
[111] Shimanovich U, Efimov I, Mason T O, et al. Protein microgels from amyloid fibril networks [J]. Acs Nano, 2015, 9 (1): 43-51.
[112] Williams R J, Hall T E, Glattauer V, et al. The in vivo performance of an enzyme-assisted self-assembled peptide/protein hydrogel [J]. Biomaterials, 2011, 32 (22): 5304-5310.
[113] Hu B, Shen Y, Adamcik J, et al. Polyphenol-binding amyloid fibrils self-assemble into reversible hydrogels with antibacterial activity [J]. Acs Nano, 2018, 12 (4): 3385-3396.
[114] Briuglia M -L, Urquhart A J, Lamprou D A. Sustained and controlled release of lipophilic drugs from a self-assembling amphiphilic peptide hydrogel [J]. International Journal of Pharmaceutics, 2014, 474 (1-2): 103-111.
[115] Krysmann M J, Castelletto V, Kelarakis A, et al. Self-assembly and hydrogelation of an amyloid peptide fragment [J]. Biochemistry, 2008, 47 (16): 4597-4605.
[116] Collier J H, Hu B H, Ruberti J W, et al. Thermally and photochemically triggered self-assembly of peptide hydrogels [J]. Journal of the American Chemical Society, 2001, 123 (38): 9463-9464.
[117] Nystrom G, Fong W -K, Mezzenga R . Ice-templated and cross-linked amyloid fFibril aerogel scaffolds for cell growth [J]. Biomacromolecules, 2017, 18 (9): 2858-2865.
[118] Nystroem G, Fernandez-Ronco M P, Bolisetty S, et al. Amyloid templated gold aerogels [J]. Advanced Materials, 2016, 28 (3): 472-478.
[119] Tang Z, Chen Q, Chen F, et al. General principle for fabricating natural globular protein-based double-network hydrogels with integrated highly mechanical properties and surface adhesion on solid surfaces [J]. Chemistry of Materials, 2019, 31(1): 179-189.
[120] Li C, Alam M M, Bolisetty S, et al. New biocompatible thermo-reversible hydrogels from PNiPAM-decorated amyloid fibrils [J]. Chemical Communications, 2011, 47 (10): 2913-2915.
[121] Blatz T J, Fry M M, James E I, et al. Templating the 3D structure of conducting polymers with self-assembling peptides [J]. Journal of Materials Chemistry B, 2017, 5 (24): 4690-4696.
[122] Sano K -I, Kawamura R, Tominaga T, et al. Thermoresponsive microtubule hydrogel with high hierarchical structure [J]. Biomacromolecules, 2011, 12 (5): 1409-1413.
[123] Tseng P, Napier B, Zhao S, et al. Directed assembly of bio-inspired hierarchical materials with controlled nanofibrillar architectures [J]. Nature Nanotechnology, 2017, 12 (5): 474-480.
[124] Li I C, Hartgerink J D. Covalent capture of aligned self-assembling nanofibers [J]. Journal of the American Chemical Society, 2017, 139 (23): 8044-8050.
[125] Zhang S, Greenfield M A, Mata A, et al. A self-assembly pathway to aligned monodomain gels [J]. Nature Materials, 2010, 9 (7): 594-601.
[126] Kamada A, Mittal N, Soderberg L D, et al. Flow-assisted assembly of nanostructured protein microfibers [J]. Proceedings of the National Academy of Sciences of the United States of America, 2017, 114 (6): 1232-1237.
[127] Zhang S G. Fabrication of novel biomaterials through molecular self-assembly [J]. Nature Biotechnology, 2003, 21 (10): 1171-1178.
[128] Zhou X -M, Shimanovich U, Herling T W, et al. Enzymatically active microgels from self-assembling protein nanofibrils for microflow chemistry [J]. Acs Nano, 2015, 9 (6): 5772-5781.
选自:韩祥生博士论文
最后于 2019-6-13
被lchaoxu编辑
,原因: